经常碰到需要计算一组DNA序列的一致性序列,比如去除测序数据中的PCR错误,最简单的方法就是通过计算它们之间的一致性序列。
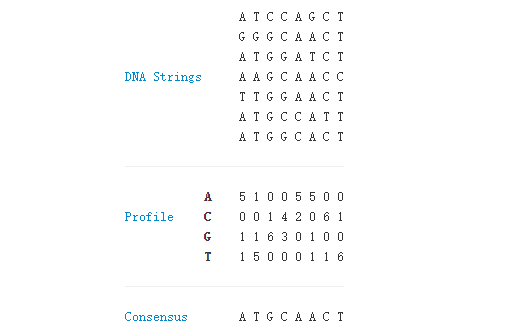
计算一致性序列,通常借助一个中间矩阵,如上图的Profile。我们可以沿着序列延伸的方向,计算每一个位点的A、C、G、T含量,从而得到一个用于计数的Profile矩阵,然后每一个位置,计数最多的碱基,就加入一致性序列中。
给定: 一个FASTA文件,其中有不超过10条,长度相等的DNA序列。
需得: 这些序列的一致性序列,以及它们的profile矩阵(可能有多条一致性序列,返回任意一条就可以了)。
示例数据
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
>Rosalind_1
ATCCAGCT
>Rosalind_2
GGGCAACT
>Rosalind_3
ATGGATCT
>Rosalind_4
AAGCAACC
>Rosalind_5
TTGGAACT
>Rosalind_6
ATGCCATT
>Rosalind_7
ATGGCACT
|
示例结果
1
2
3
4
5
|
ATGCAACT
A: 5 1 0 0 5 5 0 0
C: 0 0 1 4 2 0 6 1
G: 1 1 6 3 0 1 0 0
T: 1 5 0 0 0 1 1 6
|
Python实现
Consensus_and_Profile.py
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
|
import sys
import pysam
def consensus(infasta):
# Create profile matrix
base = 'ACGT'
profile = []
with pysam.FastxFile(infasta) as fh:
for r in fh:
if not profile:
profile = [[0] * len(r.sequence) for x in base]
for i,b in enumerate(r.sequence):
profile[base.index(b)][i] += 1
# Get consensus string
cons = ''
for i in range(len(profile[0])):
count = { b:profile[base.index(b)][i] for b in base }
cons += max(count.items(), key=lambda x: x[1])[0]
return cons, profile
def test():
cons, profile = consensus('rosalind_cons_test.txt')
#cons, profile = consensus('rosalind_cons.txt')
print(cons)
for i,base in enumerate('ACGT'):
line = ' '.join(['%s:'%base] + [str(n) for n in profile[i]])
print(line)
return cons == 'ATGCAACT'
if __name__ == '__main__':
test()
|
学生信欢迎关注我的知乎,请“点赞”为爱发电,只“收藏”不点赞都是耍流氓。
文章作者
公众号:简说基因, 知乎:简佐义
上次更新
2020-12-07
许可协议
本站文章皆为原创,转载需注明作者及文章链接,谢谢!