扩增子测序在临床基因检测中有广泛应用,合理的Panel设计非常重要,而Panel设计最终要落地,精心设计引物就是重中之重了。
本文通过公开资料整理而成,志在让外行了解一些引物设计的基础知识,深入研究请参考专业文献。本文仅供学习参考,不构成任何具体建议。
背景:DNA热力学
DNA热力学是指温度影响双链DNA(double-stranded DNA,dsDNA)的核酸结构。这部分知识是难的,但如果不深入了解,引物设计过程中可能会犯许多错误。
一些基础概念:
杂交: 正常情况下,寡核苷酸、DNA或RNA会绑定到其互补序列,互补序列的碱基之间通过氢键连接,最常见的是,核酸碱基对以A = T和G≡C的方式成对形成,其中后者更稳定(由于氢键数较多)。
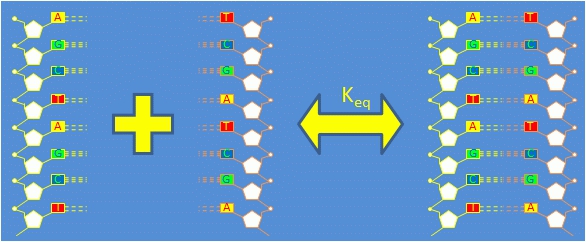
变性:又称DNA解链或融化,是DNA双链因为加热温度升高或者化学物质的诱导变成单链的过程。我们把DNA双链解开一半时所需要的温度称为融点(Melting temperature, Tm)。Tm依赖于DNA分子的长度及其特定的核酸序列组成。
复性:又称退火。单链DNA或RNA与互补的探针或引物结合形成配对的双链核苷酸的过程。
关于PCR的7个谬误及改进办法
Myth 1: PCR Nearly Always Works and Design Is Not that Important
谬误1:PCR随便弄就行了,反正都能工作。
有些时候或许确实如此,尤其是一重PCR的时候,但如果是多重PCR就必须要精心设计了。
Myth 2: Different Methods for Predicting Hybridization Tm Are Essentially Equivalent in Accuracy
谬误2:随便一种方法预测寡核苷酸的Tm值准确性都差不多。
计算Tm最简单的公式是:
Tm = 4(G + C) + 2(A + T)
这个等式忽略了许多重要信息:Tm值依赖于链的浓度,盐分浓度以及碱基序列。这个公式得到的Tm通常比实验测得的高15℃。因此并不推荐使用,一个更好的公式是:
Tm = 81.5C + 16.6 × log10[Na+]+0.41(%G+C)–0.63(%formamide)–600/L
其中L是双链DNA的长度。
Myth 3: Designing Forward and Reverse Primers to Have Mathing Tm's Is the Best Strategy to Optimize for PCR
谬误3:很多老手都相信这一点:优化PCR设计时,最好让上下游的引物都具有接近的Tm。
许多软件也是基于这一策略设计的。但这是有缺陷的,用ΔG°比Tm要好。
Myth 4: “Primer Dimer” Artifacts Are Due to Dimerization
谬误4:引物二聚体只是引物的二聚化。
常见导致引物二聚体的因素有:3'端互补,5'端或中间序列互补,高循环数(>35),掺入外源基因组DNA,引物结合靶DNA的效率低,使用具有校正活性的DNA聚合酶(Pfu等)因3'外切活性可能使原本不匹配的3'端互补。
Myth 5: A BLAST Search Is the Best Method for Determing the Specificity of a Primer
谬误5:BLAST搜索是检验引物特异性的最佳方法。
事实上,热力学参数比序列相似性能够更好地预测引物特异性。
Myth 6: At the End of PCR, Amplification Efficiency Is Not Exponential Because the Primers or NTPs Are Exhausted or the Polymerase Looses Activity
谬误6:PCR平台期的出现是因为引物或dNTP耗尽,或者酶失去活性。
实则不然,真正导致平台期的原因是积累的dsDNA产物的抑制,这已经得到了实验证明。
Myth 7: Multiplex PCR Can Succeed by Optimization of Individual PCRs
谬误7:可以通过优化单重PCR来优化多重PCR。
设计良好的单重PCR确实有助于开发多重PCR,但如果只使用这种策略则过于简单。还需要使用软件而不是人工来协助设计。
PCR引物设计原则
PCR反应中有两条引物,即5′端引物和3′引物。设计引物时以一条DNA单链为基准(常以信息链为基准),5′端引物与位于待扩增片段5′端上的一小段DNA序列相同;3′端引物与位于待扩增片段3′端的一小段DNA序列互补。
1. 引物设计基本原则
- 引物长度:18-26bp,可放宽至18-30bp(有研究表明超过30bp再增加引物长度意义不大);
- 引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C 过多易出现非特异条带。ATGC最好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列存在;
- 引物Tm:54-58℃,可放宽至52~62℃,GC%>60%可进一步放宽;
- 扩增子Tm:>92℃;
- 3'端:优选三联体WSS、SWS、TTS,二连体GC,单体S,尽量规避WWW、CGW、GGG、CG;
- 引物与非特异扩增区的序列的同源性不要超过70%,引物3′末端连续8个碱基在待扩增区以外不能有完全互补序列,否则易导致非特异性扩增;
- 末端2bp最好为GC;
- 引物的5′端可以修饰。如附加限制酶位点,引入突变位点,用生物素、荧光物质、地高辛标记,加入其它短序列,包括起始密码子、终止密码子等;
- 其他:避免3'端8bp及以上序列与模板多位点互补,尽量避免上下游3'端4bp及以上反向互补,尽量避免内部回文。
基于上述原则,怎么在实践中应用呢?这要分两种情况: (1) 基因克隆PCR引物设计
这种情况我们往往并没有太多选择,只能从ATG开始,从TAA等结束,什么GC%、二聚体和错配,可能根本由不得我们。既然起点定了,那只能通过改变长度来匹配最优设计要求了。 (2) 鉴定PCR引物设计
我们只需要确认一段DNA序列上的一部分,起点是相对的,我们可以在整个序列范围内搜索,引物设计的灵活性大大提高,当然搜索时间也要增加。
2. 引物设计软件
- Primer Premier5.0 (自动搜索)
- vOligo6 (引物评价)
- vVector NTI Suit
- vDNAsis
- vOmiga
- vDNAstar
- vPrimer3 (在线服务)
- ThermoBLAST
参考文献
- PCR Primer Design, 《Methods In Molecular Biology》第402卷
- PCR Primer Design来自于2009年第3卷(doi:doi:10.1101/pdb.ip65)
- Bioinformatic tools and guideline for PCR primer design(10.5897/AJB2003.000-1019)
- 临窗听风雨:PCR引物设计大法
- 百度百科:聚合酶链式反应
从零开始学PCR系列文章:
觉得内容不错,请点个赞吧! 日拱一卒,功不唐捐,run run run。。。